Sterile API Manufacturing
Curia provides sterile APIs for over 300 clients in more than 30 countries around the world. Our customers trust us because of our excellent track record of regulatory compliance and quality assurance.
There are several different methods which can be used to sterilize APIs, such as the terminal ones that are heat (dry or wet) and radiation. Curia offers a technology that is used in situations where the terminal ones do not guarantee the stability of the API, neither a good impurity profile. This is the aseptic processing, working under closed systems design (as isolators) throughout the entire process to ensure sterility and to minimize the risk of contamination.
Curia is uniquely positioned to handle the most complicated challenges of manufacturing
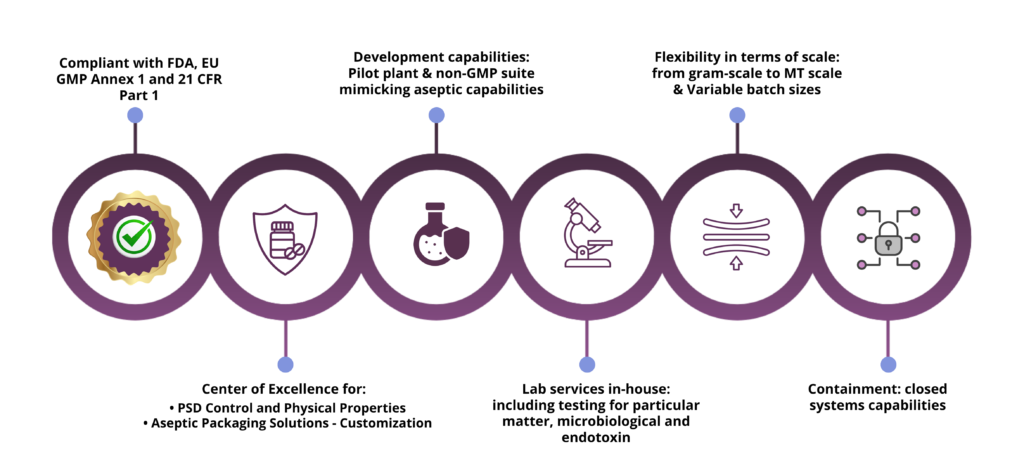
With over 20 years of experience, Curia is here to help you identifying best conditions and parameters to perform Aseptic API sterilizing filtration, with three different manufacturing suites available, and following a four-step strategy for successful sterile API manufacturing:
Stage One: Dissolution and Sterile Filtration

- Sterile API manufacturing starts with sterilization of the product by dissolving the product to filter it through a sterile 0.22 microns filter. This is where selection of the filter is critical.
- Once the sterile filtration is finished, the integrity of the filter must be checked as part of the quality assurance process.
Stage Two: Crystallization

- This stage is critical to achieve the physical properties of the product, such as crystal shape, polymorphic and particle size.
- We have a variety of methods to achieve this, including adding anti-solvent where the product is not solvable, cooling (in cases where the solvability is different depending on the temperature), distillation of the dissolving solvent obtaining a saturated concentrator and using a sterile seeding.
Stage Three: Filtration and Drying

- The goal here is to separate the API from the solvent and dry the product to remove the voluntary impurities until we achieve the desired purification.
- At this stage, we are especially careful to avoid the degradation of the product from conditions such as temperature and moisture.
- Operations are carried out through isolator to avoid contact between people and the product.
Stage Four: Physical Treatment and Packaging

- If necessary, the dried material can be milled or micronized to reduce particle size.
- API is packaged according to the customer’s needs, using a range of customizable solutions.
Apart from the main process, we have secondary processes that we use to minimize risk in sterile API manufacturing. These include operator training to prevent contamination during handling, environmental monitoring to check all conditions and operations are right to assure the safety conditions, pressure cascade control to avoid contamination between rooms and particle control strategies to avoid foreign particles.


Frequently Asked Questions
What are the key principles for sterile product manufacture in the pharmaceutical industry?
The manufacture of sterile products is a complex activity that requires specific controls and measures to ensure the
quality of products manufactured.
The key principles of sterile product manufacturing include minimizing the risk of microbial, particulate, and endotoxin
contamination through appropriate facility and equipment design, qualified personnel, validated processes, and robust
monitoring systems. Quality Risk Management (QRM) and a documented Contamination Control Strategy (CCS) should
be applied throughout all operations.
Which types of drug product formulations could request a sterile API?
Sterile APIs may be required for several types of pharmaceutical formulations, including:
- Injectables
- Inhalation products
- Implants
- Ophthalmic formulations
What methods are commonly used to sterilize active pharmaceutical ingredients (APIs)?
Active pharmaceutical ingredients (APIs) can be sterilized using several terminal sterilization methods, including dry
heat, moist heat, radiation, and other validated approaches. These methods are suitable when the API remains stable
under the required sterilization conditions and its molecular structure and impurity profile are not adversely affected.
Where terminal sterilization of the APIs is not feasible, sterilization by filtration followed by aseptic processing under
validated and controlled conditions is the option of choice.
What is the best alternative approach when terminal sterilization is not feasible?
When terminal sterilization is not feasible, sterile filtration followed by aseptic processing is the primary alternative. This specialized manufacturing approach ensures sterility by preventing contamination throughout the full production workflow and process rather than sterilizing the product at the final stage.
What types of contamination does aseptic processing aim to prevent?
Aseptic processing is intended to prevent microbial, particulate, and endotoxin contamination. These controls help
ensure that the final API meets the sterility requirements necessary for use in sterile pharmaceutical formulations.
How can sterility be ensured during aseptic processing?
Sterility during aseptic processing is ensured through the application of Quality Risk Management (QRM) across all
stages of aseptic API production. This includes facility design, equipment qualification, process and controls design,
system implementation, and operational procedures. QRM provides a structured framework for identifying, assessing,
and controlling risks associated with sterile manufacturing. It supports consistent process performance and helps
ensure that the final API meets stringent sterility and quality requirements.
What are the global regulatory and compliance standards?
Aseptic capabilities should comply with the latest international regulatory requirements. The principal applicable
standards include:
- EU GMP Annex 1
- FDA Guidance for Industry: Sterile Drug Products. Current good Manufacturing Practices
- FDA guidelines (21 CFR Part 11)
- ISO standards, including ISO 13408 and ISO 14644
- PDA technical reports (TR), including TR 28 and TR 27
- USP <1116>
Together, these frameworks provide general guidance for the design and control of facilities, equipment, systems, and
procedures used in sterile product manufacturing, while reinforcing the application of Quality Risk Management (QRM)
principles.
What are the key requirements for premises and cleanroom design for aseptic API processing?
Key requirements for premises and cleanroom design include the following:
- Controlled environments: Sterile manufacturing should be carried out in cleanrooms equipped with HEPA-filtered air to prevent microbial ingress. Appropriate airlocks and physical separation are required to maintain environmental control. Cleanrooms are classified into four grades (A–D), each with defined requirements.
- Closed systems and automation: Manufacturing systems should be designed to minimize human intervention. Barrier technologies such as RABS and isolators, together with automation, are recommended to reduce contamination risk.
- Equipment and utilities: Equipment should support effective cleaning and minimize contamination risk. Critical utilities, including water, gases, and steam, must be controlled, qualified, and periodically monitored.
What are the key requirements for aseptic API processing control?
Effective aseptic API processing control depends on the following elements:
- Personnel requirements: Only qualified and trained personnel should be permitted to enter cleanrooms. Gowning and hygiene requirements must be defined according to cleanroom grades. Continued training, qualifications, and routine monitoring should be supported by formal procedures for disqualification and retraining when needed.
- Environmental monitoring: Comprehensive monitoring of viable and non-viable particles is required, with alert and action limits established according to cleanroom grade. Routine trend analysis should be used to identify shifts that may affect process control. Cleanrooms should also be qualified and periodically requalified.
- Process monitoring: Robust validated procedures are required to demonstrate control of aseptic operations. These should include media fill runs designed to simulate worst case conditions. Periodic verification of the effectiveness of the controls in place for aseptic processing should include Aseptic Process Simulation (APS), also referred to as media fill. APS should be performed as part of the initial validation and periodically executed. Additional controls, such as sterilizing filter integrity testing and glove integrity monitoring, should also be included.
- Quality control (QC): Quality control should include expertise in microbiology, sterility assurance, and particulate control. Bioburden and endotoxin testing must be performed at defined stages. Sterility testing is the final verification step, but it should not be considered the sole assurance of sterility. Personnel, Environmental and process monitoring results and trend data should be reviewed as part of batch release.
- Adherence to the Pharmaceutical Quality system (PQS): the manufacturer’s PQS should encompass and address the specific requirements of sterile product manufacture and ensure that all activities are effectively controlled so that the risk of microbial, particulate and endotoxin/pyrogen contamination is minimized in sterile products.
Why consider a CDMO for your sterile API requirements?
Aseptic processing for sterile APIs is a highly controlled, complex activity that relies on purpose-built facilities, qualified personnel, validated processes, and robust monitoring to minimize risks of microbial, particulate, and endotoxin contamination. By leveraging validated aseptic processes, routine environmental and process monitoring, and
integrated microbiological expertise, a CDMO helps ensure consistent sterility assurance aligned with global regulatory expectations, while reducing the need for significant internal investment and accelerating timelines to clinical or commercial supply.